This section lists peer-reviewed publications related to HAL’s MD package.
Please send suggestions for papers that should be included to
info@halmd.org along with a short description and a
graphical illustration (if available).
The paper describes the essential MD simulation algorithms and their
implementation for the GPU. Please refer to it in all publications based on
or linked to HAL’s MD package.
The fluid flow in nanopores depends on the interaction with the pore
surfaces, but also on their atomic-scale morphology. Matching the
molecular structures of pore walls and fluid minimises the surface slip,
with crystalline and amorphous walls producing distinct flow resistances.
Thus, surface morphology offers a means to control surface slip in
non-equilibrium molecular dynamics (NEMD) simulations and emphasize the
need for molecular-scale models to accurately capture fluid dynamics in
nanoporous materials.
Liquid flow in a slit-shaped pore with amorphous walls
(courtesy of G. Marcelli)¶
Adaptive resolution simulation of open systems out of equilibrium
The Adaptive Resolution Simulation (AdResS) technique permits molecular
dynamics simulations with open boundaries by a transparent coupling to
particle reservoirs. Translating the theoretical model of Bergmann and
Lebowitz for open systems out of equilibrium to AdResS yields NEMD
simulations of the unconstrained (NVE) dynamics of fluids with open
boundaries in a thermal gradient. A pressure gradient with an ensuing mass
flux is maintained if a permeable mebrane is used to resist the fluid flow.
Thermal isolation of the fluid and viscous dissipation cause a rise in
fluid temperature, consistent with fluid mechanics.
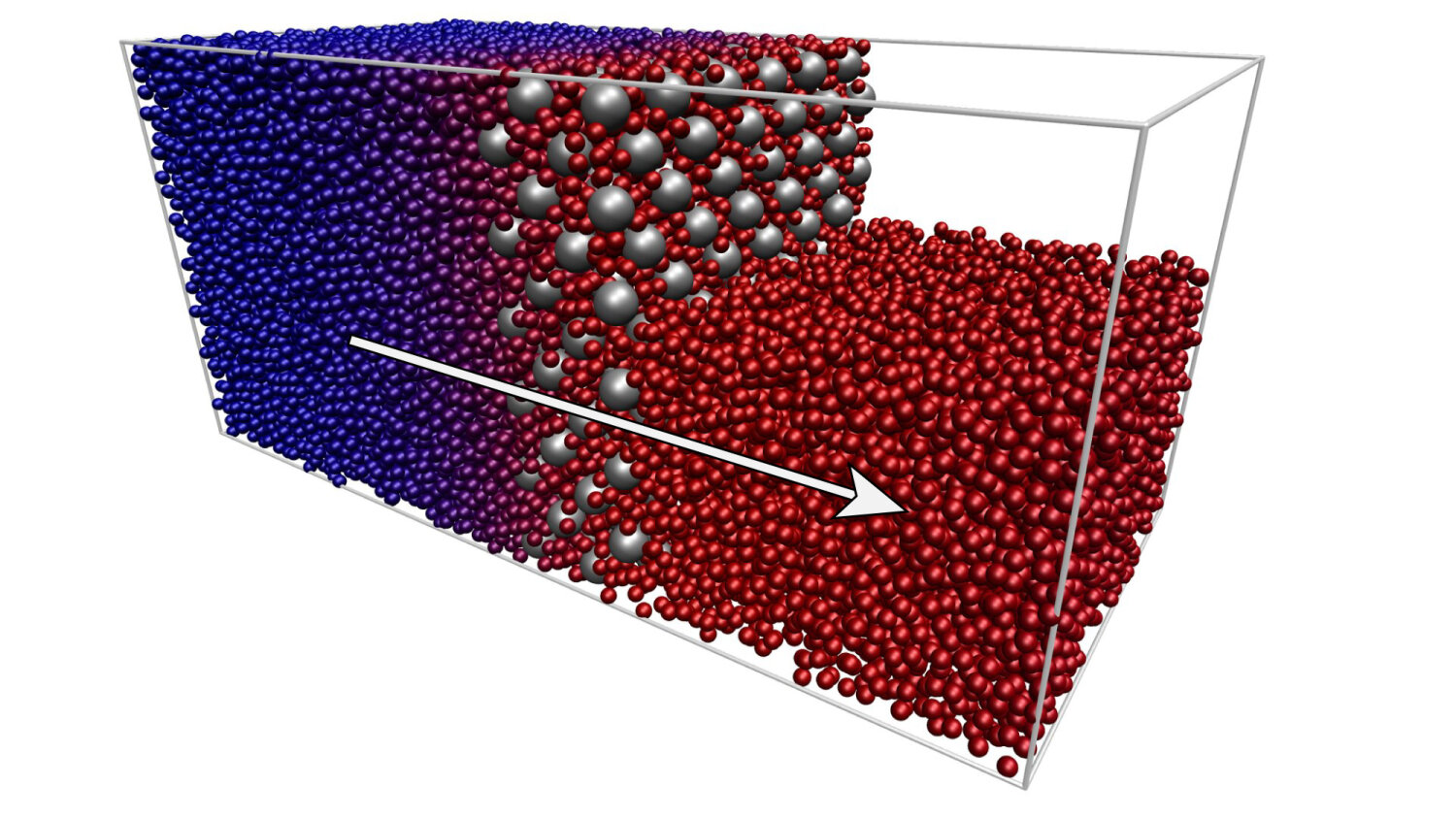
Liquid permeation of an arrangement of obstacles via boundary-driven
NEMD simulations (courtesy of F. Höfling).¶
Frequency-dependent transport coefficients in viscous liquids
How friction in liquids emerges from conservative forces between atoms is a
crucial parameter for dynamic processes in liquid matter and soft
materials. Using energy-conserving molecular dynamics simulations of simple
and complex liquids, frequency-resolved coefficients of molecular friction
and shear viscosity are obtained from high-precision data for response
functions, covering several decades in time. This approach bridges
hydrodynamic long-time anomalies and a frictionless high-frequency regime.
Combining simulation data with theory shows that the friction felt by a
single molecule occurs abruptly below a certain frequency.
At the scale of a picosecond, the smooth, but irregular atomic
trajectories lead to the rapid onset of dissipation and friction
(journal cover in October 2020)¶
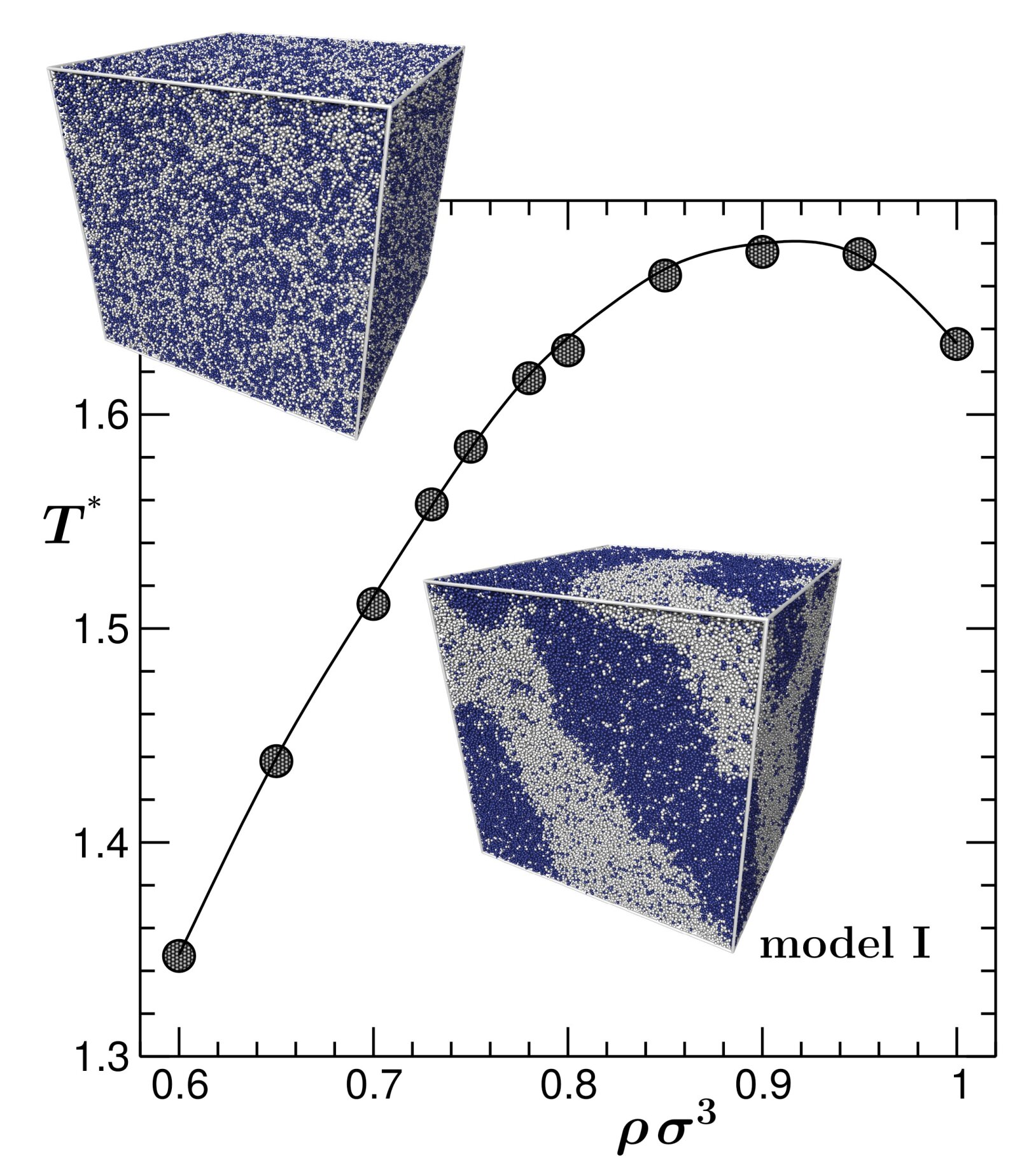
Critical dynamics and surface adsorption of symmetric binary fluids
Binary liquids phase separate at certain thermodynamic conditions.
Continous demixing transitions occur along the so-called λ-line, where
critical opalescence, long-ranged correlations, and scaling behaviour are
observed. Confinement of the mixture to a slit pore leads to surface
enrichment of one component with corresponding adsorption profiles
exhibiting critical scaling. Diverging length and time scales challenge the
simulation of these phenomena and require substantial computational
resources. For a family of binary Lennard-Jones liquids, the partial
structure factors, mutual or inter-diffusion constants and the shear
viscosity were obtained in molecular dynamics simulations.
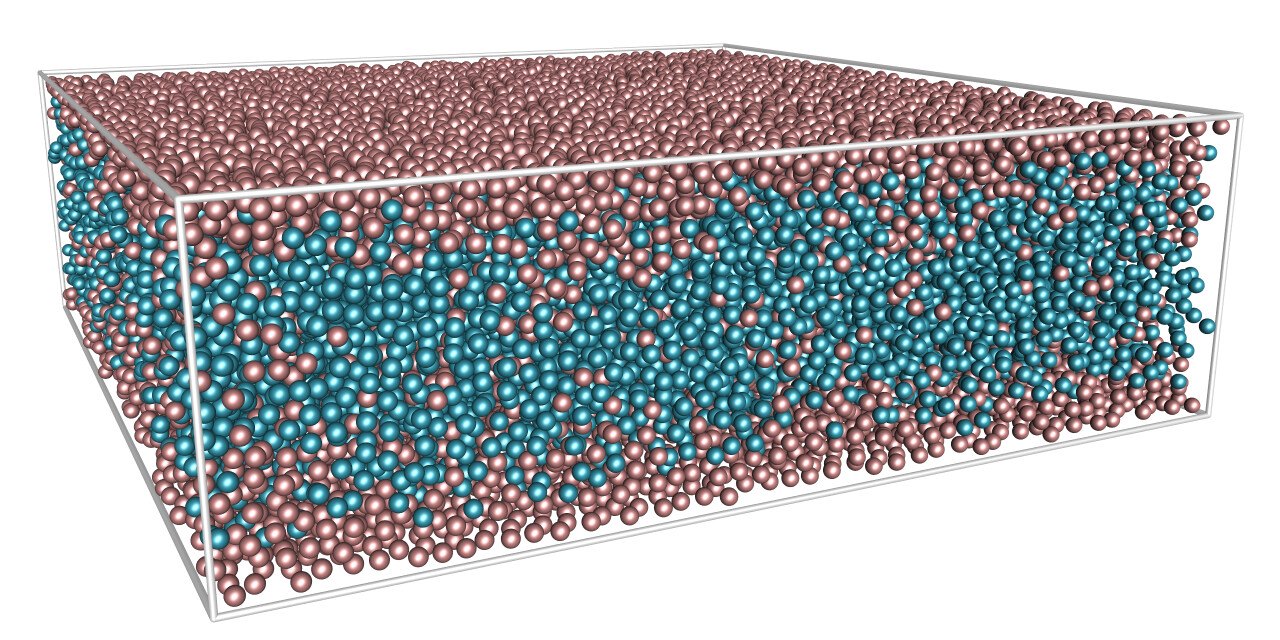
Left: adsorption of a near-critical binary liquid to a slit pore.
Right: λ-line of demixing transitions of a symmetric binary liquid
(courtesy of S. Roy)
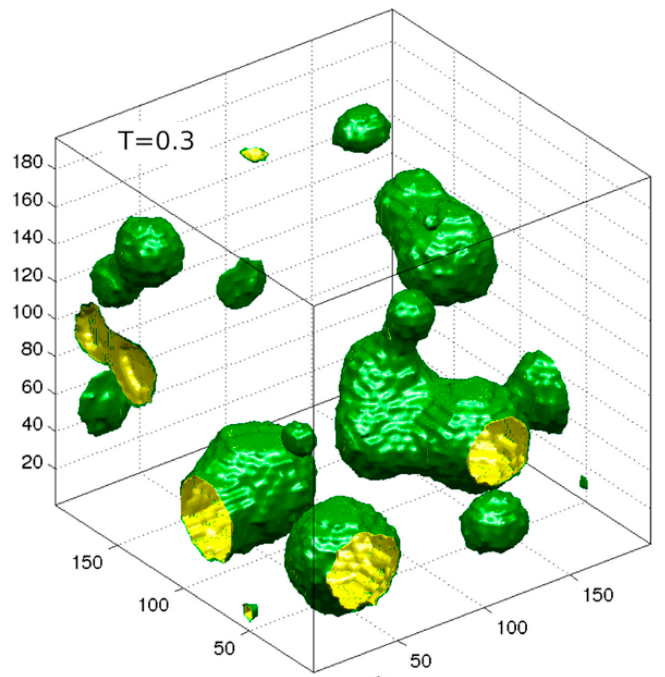
Cavitation in glass-like, amorphous solids
Continous expansion of a dense, amorphous solid leads to the formation of
cavities and, eventually, failure. The picture depicts long-lived bubbles,
which exist near the thermodynamic coexistence of gas phase (yellow) and
glassy state (green). The study is based on extensive molecular dynamics
simulations for system sizes of up to one million particles over long time
spans.
Cavitation bubbles in the expansion of an amorphous solid
(courtesy of P. Chaudhuri)¶
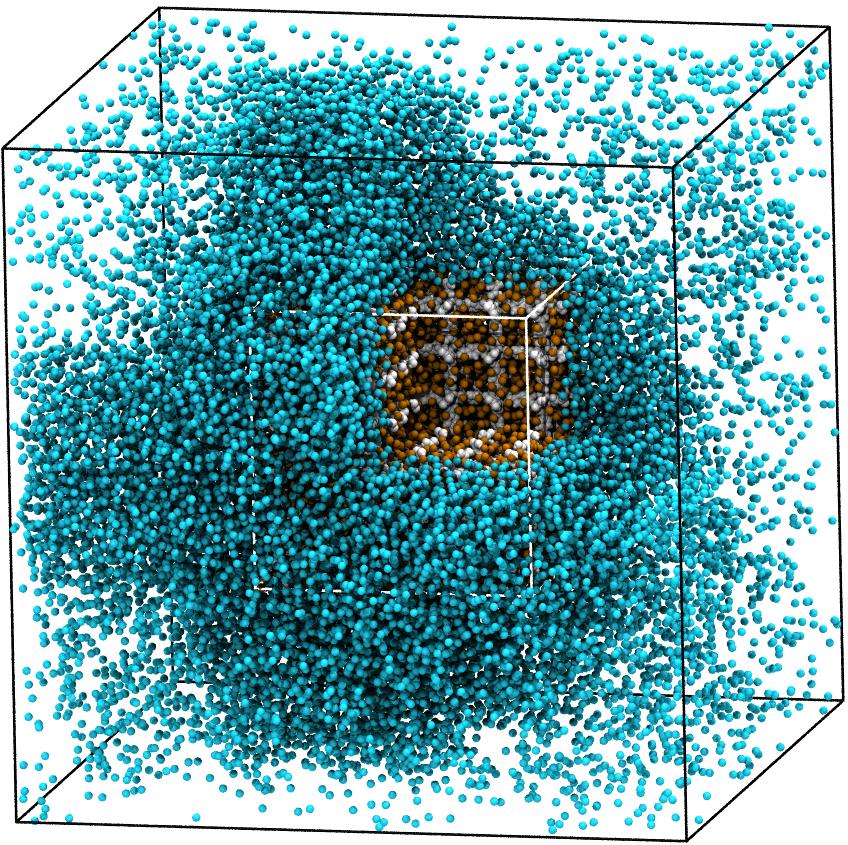
Adsorption kinetics of methane in metal-organic frameworks
The high porosity and large surface area of metal-organic frameworks (MOFs)
makes them interesting for technical applications. In contact with gaseous
methane (CH₄), one observes that the methane condenses in and around the
MOF already for unusually low gas pressure. Molecular dynamics simulations
give insight into the kinetics of the adsorption process into a single
grain of IRMOF-1.
Adsorption of an IRMOF-1 grain supsended in a CH₄ droplet
(courtesy of N. Höft)¶
Mesoscopic structure of liquid–vapour interfaces
At the molecular scale, liquid–vapour interfaces are broadened and
roughened by thermally excited capillary waves. These fluctuations give
rise to a divergence of the interfacial structure factor at small
wave-numbers; the latter being accessible to grazing-incidence small-angle
X-ray scattering (GISAXS) experiments. The paper discusses deviations from
the classical theory and relies on extensive simulations of planar interfaces
using up to 445,000 Lennard-Jones particles. GISAXS intensities are
computed on the fly of the simulations.