Thermodynamics¶
This module calculates the instantaneous values of thermodynamic state variables for the particles of a given group.
-
class
halmd.observables.thermodynamics(args)¶ Construct thermodynamics module.
Parameters: - args (table) – keyword arguments
- args.group – instance of
halmd.mdsim.particle_groups - args.box – instance of
halmd.mdsim.box
-
particle_number()¶ Returns the number of particles
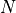 selected by
selected by args.group.
-
density()¶ Returns the number density
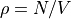 using the volume from
using the volume from
args.box.
-
kinetic_energy()¶ Returns the mean kinetic energy per particle:
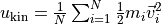 .
.
-
potential_energy()¶ Returns the mean potential energy per particle:
 ,
where
,
where 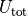 denotes the sum of external and pair
potentials.
denotes the sum of external and pair
potentials.
-
internal_energy()¶ Returns the mean internal energy per particle:
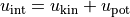 .
.
-
pressure()¶ Returns the pressure computed from the virial:
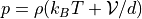 .
.
-
temperature()¶ Returns the instantaneous temperature as given by the kinetic energy:
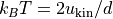 .
.
-
total_force()¶ Returns the total force:
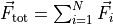 .
.
-
center_of_mass_velocity()¶ Returns the centre-of-mass velocity:
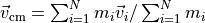 .
.
-
center_of_mass()¶ Returns the centre of mass:
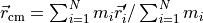 ,
where
,
where 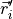 refers to absolute particle positions, i.e.,
extended by their image vectors for periodic boundary conditions.
refers to absolute particle positions, i.e.,
extended by their image vectors for periodic boundary conditions.
-
mean_mass()¶ Returns the mean particle mass:
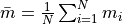 .
.
-
virial()¶ Returns mean virial per particle as computed from the trace of the potential part of the stress tensor:
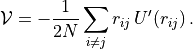
-
stress_tensor()¶ Returns the elements of the stress tensor
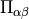 as a
vector. The first
as a
vector. The first 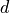 (= dimension) elements contain the diagonal
followed by
(= dimension) elements contain the diagonal
followed by 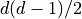 off-diagonal elements
off-diagonal elements  The stress tensor is computed as
The stress tensor is computed as![\Pi_{\alpha\beta} = \sum_{i=1}^N \left[
m_i v_{i\alpha} v_{i\beta} -
\frac{1}{2}\sum_{j \neq i} \frac{r_{ij\alpha} r_{ij\beta}}{r_{ij}} \, U'(r_{ij})
\right] \/,](../../_images/math/c4007b16996b2a9f2387ba780359edd9f0e62c34.png)
where
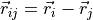 in nearest image convention.
in nearest image convention.
-
dimension¶ Space dimension
of the simulation box as a number.
-
group¶ Instance of
halmd.mdsim.particle_groupsused to construct the module.
-
writer(args)¶ Write state variables to a file.
Parameters: - args (table) – keyword arguments
- args.file – instance of file writer
- args.every (number) – sampling interval
- args.location (string table) – location within file (optional)
- args.fields (table) – data fields to be written (optional)
Returns: instance of group writer
The optional argument
locationspecifies a path in a structured file format like H5MD given as a table of strings. It defaults to{"observables", group.global and nil or group.label}.The optional table
fieldsspecifies which data fields are written. It may either be passed as an indexed table, e.g.{"pressure"}, or as a dictionary, e.g.,{p = "pressure"}; the table form is interpreted as{pressure = "pressure", …}. The keys denote the field names in the file and are appended tolocation. The values specify the data methods of thethermodynamicsmodule, i.e., all methods described above except fordimensionandgroup. The default is{"potential_energy", "pressure", "temperature", "center_of_mass_velocity"}.-
disconnect()¶ Disconnect thermodynamics writer from observables sampler.